ゴーシェ病とは?
ゴーシェ病(Gaucher disease)は、細胞内リソソーム(ライソゾーム)において加水分解酵素であるグルコセレブロシダーゼ(β-グルコシダーゼ)活性が正常値より不足あるいは欠損しているため、この酵素で本来分解されるべき糖脂質のグルコセレブロシドが肝臓、脾臓、骨髄(マクロファージ)などに蓄積する先天性代謝異常症です。
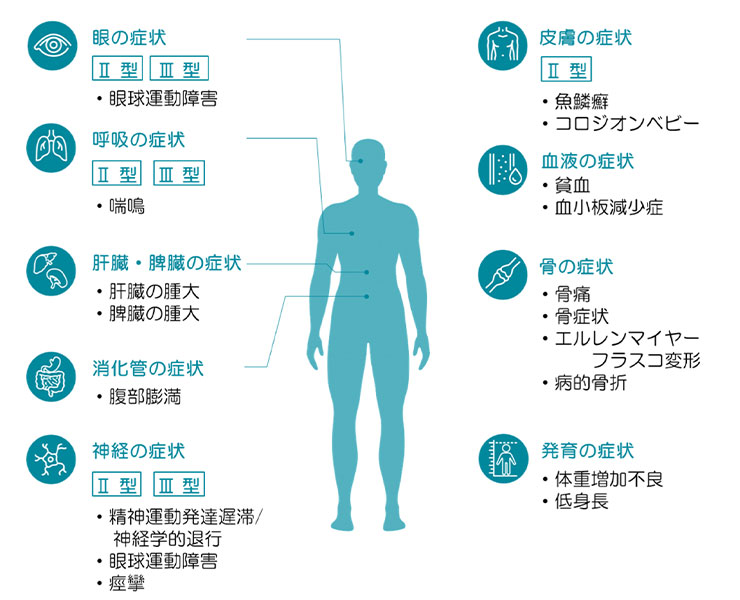
主に、肝臓、脾臓、骨髄などに蓄積し、貧血、血小板減少症、肝臓・脾臓の腫大、骨痛・骨症状などに加えて、神経症状を呈する場合があります。
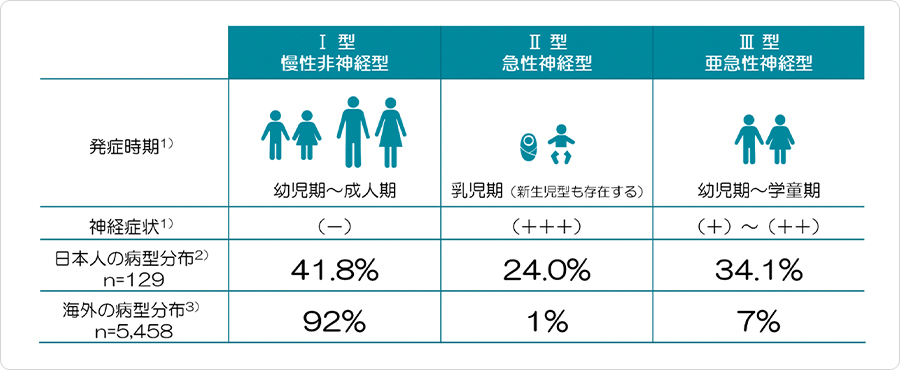
病型は、神経症状の有無や臨床症状により1型~3型に分類されており、わが国では約100名の患者さんが報告されています。
遺伝形式は常染色体潜性(劣性)遺伝です。
どんな症状が出るの?
| 肝・脾腫 | 肝臓や脾臓が腫れて大きくなります。 |
|---|---|
| 貧血、血小板減少症 | 骨髄のゴーシェ細胞が増加することによる造血能の低下や、脾腫による赤血球、血小板の破壊の亢進がみられます。 |
| 骨症状 | 骨が痛くなったり、弱くなって骨折したりします。 |
| 神経症状 | 発達遅延やけいれんをおこす人もいます。 |
症状には個人差があります。
病型は?
-
1型(非神経型)発症年齢が幼児から成人にわたり、慢性に経過します。神経症状を伴わず、肝脾腫、骨症状が主症状です。
-
2型(急性神経型)乳児期(生後3~5ヶ月頃)に発症し、著明な神経症状(精神運動発達遅延、けいれん、頸部後屈、開口困難、斜視)を伴います。胎児水腫が主症状です。
-
3型(亜急性神経型)乳幼児期に徐々に発症し、神経症状を伴いますが、2型に比べて緩徐な経過をたどります。
3型はさらに3つの亜型に分類されます。しばしば肝脾腫が初発症状となります。
どうやって診断するの?
血液を少し採取して、血液中の酵素・グルコセレブロシダーゼ(β-グルコシダーゼ)の活性を測定します。
他にも、骨髄や肝臓・脾臓の組織を少し採取してゴーシェ細胞や蓄積している糖脂質(グルコセレブロシド)の分析、グルコセレブロシダーゼ遺伝子変異を調べることがあります。
ゴーシェ病を疑う所見
- 貧血、血小板減少症
- 肝臓・脾臓の腫大
- 骨痛、骨症状
その他、アンジオテンシン変換酵素(ACE)値上昇、ゴーシェ細胞、病的骨折、エルレンマイヤーフラスコ変形などの症状があり、Ⅱ・Ⅲ型では神経症状(眼球運動障害、精神運動発達遅滞、痙攣など)があります。
どうやって治療するの?
遺伝子組換えグルコセレブロシダーゼ製剤を補充する酵素補充療法(ERT)または、グルコセレブロシドの合成を抑制する基質合成阻害療法(SRT)があります。酵素補充療法は、欠損している酵素を体外から点滴で補充し、蓄積している糖脂質を代謝・分解する治療法です。基質合成阻害療法は、既定の薬を毎日内服する治療法です。
他には、貧血、血小板減少症、脾臓の腫大、骨痛、骨症状、神経症状などへの対症療法が必要となる場合があります。
